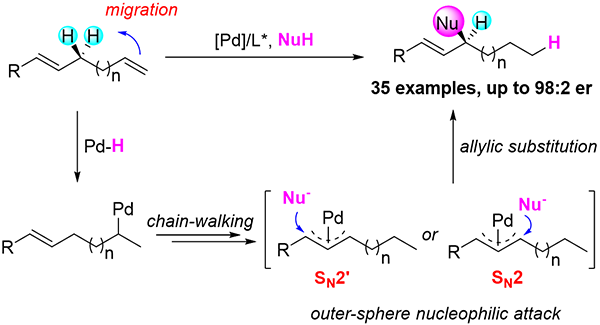
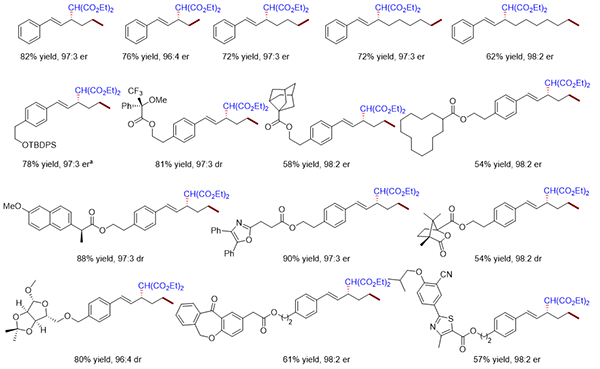
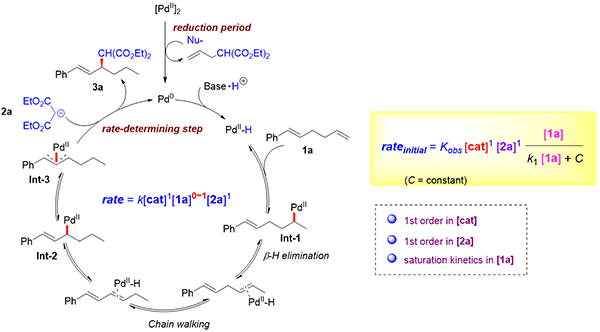
上海有機所在PdH催化的不對稱遷移烯丙基取代方面取得研究進展
發(fā)布時間:2021-09-26中科院天然產(chǎn)物有機合成化學(xué)重點實驗室| 【 大 中 小 】【打印】 【關(guān)閉】
附件下載: